Protein misfolding disorders
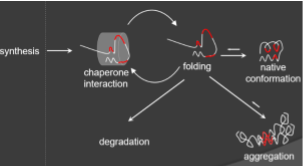
Protein misfolding is a leading mechanism in many disease pathologies. In Parkinson’s and Alzheimer’s disease certain proteins misfold and aggregate inside and outside cells whereas mutant proteins in many inborn errors of metabolism misfold and are degraded. The degree of aggregation or degradation of misfolding proteins depends on the activity of molecular chaperones and proteases that form protein quality control systems and modulates pathogenicity. We studied this in connection with fatty acid oxidation disorders and investigate diseases that are caused by mutations in a chaperone that is central for protein quality control inside mitochondria.
Examples of projects:
A zebrafish model for deficiency of the mitochondrial HSP60 chaperone The zebrafish gene encoding HSP60 was knocked out by CRISPR/cas technology and mutant embryos were examined by fluorescence microscopic analysis of myelination, effects on gene expression by RNASeq and proteomics, and targeted metabolomics.
Studies of disease association of de novo mutations De novo mutations in the gene encoding the mitochondrial HSP60 chaperone have been detected in patients with hypomyelination and developmental disorders. To corroborate the disease association of these mutations we have studied HSP60 expression and activity using patient skin fibroblasts and genetic complementation systems.
Contact: Associate Professor Peter Bross, MSc, PhD (peter.bross@clin.au.dk).